Squidpy-GPU#
Accelerated Spatial Analysis
Author: Severin Dicks Copyright scverse
Here, we explore GPU-accelerated spatial analysis using rapids-singlecell’s rsc.gr module, which mirrors the API of Squidpy.
We compute spatial autocorrelation metrics — Moran’s I and Geary’s C — to detect spatially structured genes within tissue sections.
By running these analyses on GPUs, we achieve faster computation and scalability for large spatial transcriptomics datasets, revealing biological patterns such as niche-specific expression and spatial gradients.
import squidpy as sq
import rapids_singlecell as rsc
import cupy as cp
import rmm
from rmm.allocators.cupy import rmm_cupy_allocator
rmm.reinitialize(
managed_memory=False, # Keep allocations in device memory
pool_allocator=True, # default is False
devices=0, # GPU device IDs to register. By default registers only GPU 0.
)
cp.cuda.set_allocator(rmm_cupy_allocator)
adata = sq.datasets.visium_hne_adata()
Constructing a Spatial Graph#
Before computing spatial autocorrelation, we first need to build a spatial neighbors graph,
which defines spatial relationships between cells or tissue spots.
We use Squidpy to compute the graph based on spatial coordinates stored in adata.obsm["spatial"]
%%time
sq.gr.spatial_neighbors(adata)
CPU times: user 6 ms, sys: 89 μs, total: 6.08 ms
Wall time: 5.42 ms
genes = adata.var_names
Moran’s I#
To assess spatial structure in gene expression, we compute spatial autocorrelation,
which measures how gene expression levels are spatially distributed across tissue sections.
%%time
rsc.gr.spatial_autocorr(adata, mode="moran", genes=genes, n_perms=100, use_sparse=False)
CPU times: user 396 ms, sys: 142 ms, total: 538 ms
Wall time: 533 ms
adata.uns["moranI"]
| I | pval_norm | var_norm | pval_z_sim | pval_sim | var_sim | pval_norm_fdr_bh | pval_z_sim_fdr_bh | pval_sim_fdr_bh | |
|---|---|---|---|---|---|---|---|---|---|
| Nrgn | 0.874753 | 0.000000 | 0.000131 | 0.000000 | 0.009901 | 0.000323 | 0.000000 | 0.000000 | 0.016667 |
| Mbp | 0.868723 | 0.000000 | 0.000131 | 0.000000 | 0.009901 | 0.000333 | 0.000000 | 0.000000 | 0.016667 |
| Camk2n1 | 0.866545 | 0.000000 | 0.000131 | 0.000000 | 0.009901 | 0.000296 | 0.000000 | 0.000000 | 0.016667 |
| Slc17a7 | 0.861761 | 0.000000 | 0.000131 | 0.000000 | 0.009901 | 0.000300 | 0.000000 | 0.000000 | 0.016667 |
| Ttr | 0.841988 | 0.000000 | 0.000131 | 0.000000 | 0.009901 | 0.000328 | 0.000000 | 0.000000 | 0.016667 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Slc9a8 | -0.026960 | 0.010027 | 0.000131 | 0.000327 | 0.009901 | 0.000066 | 0.017979 | 0.000610 | 0.016667 |
| Pramef8 | -0.028659 | 0.006681 | 0.000131 | 0.000011 | 0.009901 | 0.000043 | 0.012348 | 0.000024 | 0.016667 |
| Klf12 | -0.028764 | 0.006512 | 0.000131 | 0.000087 | 0.009901 | 0.000061 | 0.012052 | 0.000171 | 0.016667 |
| Gart | -0.028859 | 0.006361 | 0.000131 | 0.000384 | 0.009901 | 0.000077 | 0.011784 | 0.000713 | 0.016667 |
| Zbed4 | -0.030420 | 0.004295 | 0.000131 | 0.000092 | 0.009901 | 0.000066 | 0.008154 | 0.000181 | 0.016667 |
18078 rows × 9 columns
Geary’s C#
In addition to Moran’s I, we compute Geary’s C, another spatial autocorrelation metric.
While Moran’s I captures global spatial structure, Geary’s C is more sensitive to local differences,
highlighting regions with abrupt expression changes.
use_sparse=True Enables sparse computation, preventing full matrix densification.
This optimization is handled efficiently in the GPU kernel, significantly reducing memory usage.
Moran’s I also supports use_sparse=True, allowing for efficient computation on large datasets.
%%time
rsc.gr.spatial_autocorr(adata, mode="geary", genes=genes, n_perms=100, use_sparse=True)
CPU times: user 527 ms, sys: 40 ms, total: 567 ms
Wall time: 562 ms
adata.uns["gearyC"]
| C | pval_norm | var_norm | pval_z_sim | pval_sim | var_sim | pval_norm_fdr_bh | pval_z_sim_fdr_bh | pval_sim_fdr_bh | |
|---|---|---|---|---|---|---|---|---|---|
| Nrgn | 0.126481 | 0.000000e+00 | 0.000131 | 0.000000 | 0.009901 | 0.000337 | 0.000000e+00 | 0.000000 | 0.016733 |
| Mbp | 0.132854 | 0.000000e+00 | 0.000131 | 0.000000 | 0.009901 | 0.000300 | 0.000000e+00 | 0.000000 | 0.016733 |
| Camk2n1 | 0.135106 | 0.000000e+00 | 0.000131 | 0.000000 | 0.009901 | 0.000322 | 0.000000e+00 | 0.000000 | 0.016733 |
| Slc17a7 | 0.138819 | 0.000000e+00 | 0.000131 | 0.000000 | 0.009901 | 0.000322 | 0.000000e+00 | 0.000000 | 0.016733 |
| Ttr | 0.156155 | 0.000000e+00 | 0.000131 | 0.000000 | 0.009901 | 0.000343 | 0.000000e+00 | 0.000000 | 0.016733 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| AC166344.1 | 1.079271 | 2.059686e-12 | 0.000131 | 0.421383 | 0.049505 | 0.000050 | 7.064124e-12 | 0.436020 | 0.071936 |
| Gm14091 | 1.083541 | 1.372236e-13 | 0.000131 | 0.424654 | 0.148515 | 0.001859 | 4.932845e-13 | 0.438104 | 0.189007 |
| Slc6a5 | 1.087126 | 1.265654e-14 | 0.000131 | 0.447575 | 0.405941 | 0.000377 | 4.722497e-14 | 0.455640 | 0.428381 |
| Soat2 | 1.093646 | 1.110223e-16 | 0.000131 | 0.436112 | 0.326733 | 0.000165 | 4.432556e-16 | 0.446482 | 0.361973 |
| Bmp8b | 1.125921 | 0.000000e+00 | 0.000131 | 0.385002 | 0.099010 | 0.000045 | 0.000000e+00 | 0.413767 | 0.133247 |
18078 rows × 9 columns
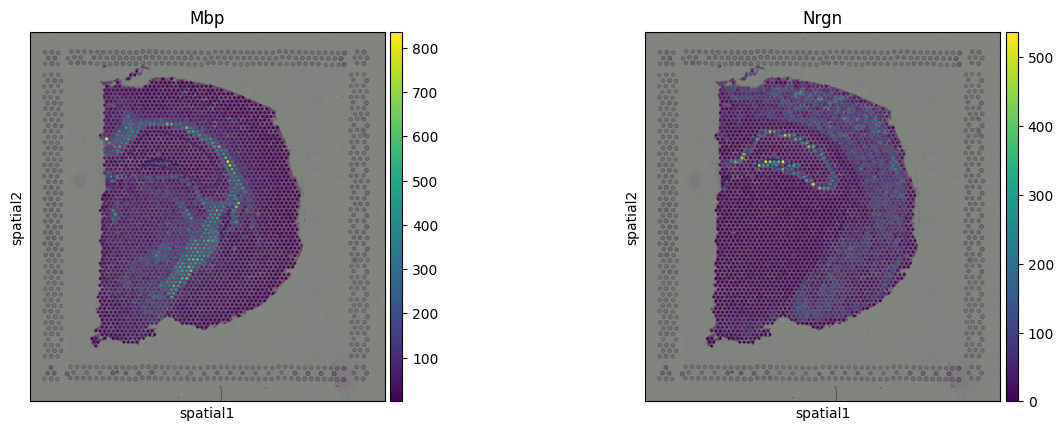
To explore spatial patterns in gene expression, we use Squidpy’s spatial scatter plot,
which overlays expression values onto the spatial coordinates of cells or tissue spots.
We visualize the expression of Mbp (myelin-associated) and Nrgn (neuronal marker):
sq.pl.spatial_scatter(adata, color=["Mbp", "Nrgn"])