Decoupler-GPU#
Accelerated Transcriptional Regulatory Analysis
Author: Severin Dicks Copyright scverse
Here, we explore the updated Decoupler functions for inferring transcriptional regulatory activity, leveraging the GPU-accelerated implementation from rapids-singlecell.
By running these analyses on GPUs, we achieve faster computation and scalability for large single-cell datasets. We will apply Decoupler methods to estimate transcription factor (TF) activity and pathway activity, utilizing curated regulatory networks such as CollecTRI.
import rapids_singlecell as rsc
import scanpy as sc
import cupy as cp
import pandas as pd
import anndata as ad
import decoupler as dc
import rmm
from rmm.allocators.cupy import rmm_cupy_allocator
rmm.reinitialize(
managed_memory=False, # Keep allocations in device memory
pool_allocator=True, # default is False
)
cp.cuda.set_allocator(rmm_cupy_allocator)
ℹ️ Note: The dataset used in this notebook is generated in 01_basic_workflow.ipynb.
adata = ad.read_h5ad("h5/dli_decoupler.h5ad")
adata = adata.raw.to_adata()
rsc.get.anndata_to_GPU(adata)
CollecTRI network#
CollecTRI is a comprehensive resource containing a curated collection of TFs and their transcriptional targets compiled from 12 different resources. This collection provides an increased coverage of transcription factors and a superior performance in identifying perturbed TFs compared to other literature based GRNs such as DoRothEA. Similar to DoRothEA, interactions are weighted by their mode of regulation (activation or inhibition).
collectri = dc.op.collectri(organism="human",license="commercial")
Transcription Factor Activity Inference with ULM#
To infer transcription factor (TF) activity, we use the Univariate Linear Model (ULM) from Decoupler.
ULM estimates the regulatory influence of TFs on gene expression by fitting a linear model for each TF-gene interaction.
We apply ULM using the Dorothea regulatory network retrieved earlier.
%%time
rsc.dcg.ulm(data=adata, net=collectri, verbose = True, raw=False, bsize=10000, tmin=3)
CPU times: user 16.4 s, sys: 610 ms, total: 17 s
Wall time: 17 s
score = dc.pp.get_obsm(adata=adata, key="score_ulm")
sc.pl.embedding(score,basis="X_umap_harmony", color=["cell_type", "GATA3"], cmap='RdBu_r',ncols=1)
sc.pl.violin(score, keys=['GATA3'], groupby='cell_type', rotation=90, size = 0)
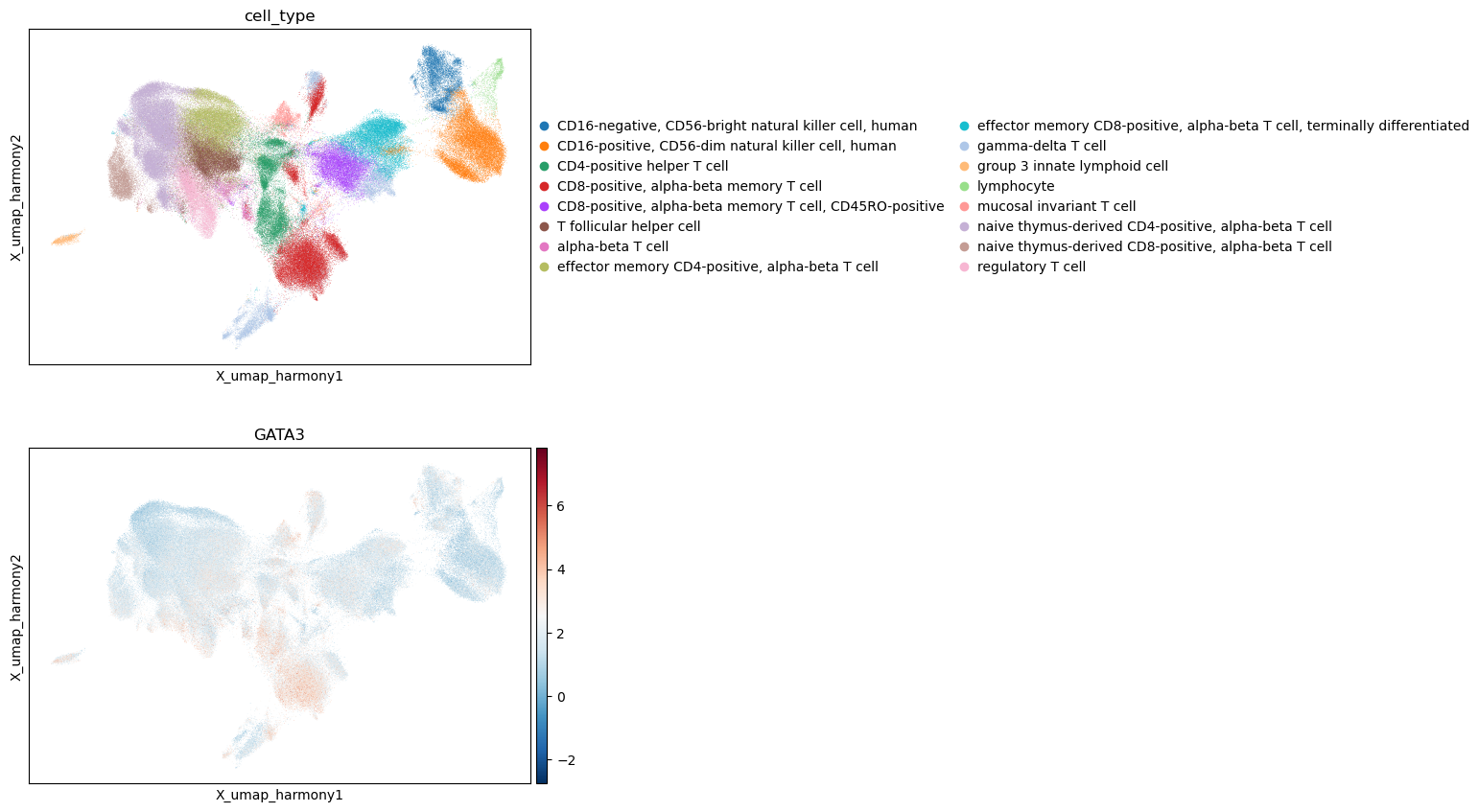
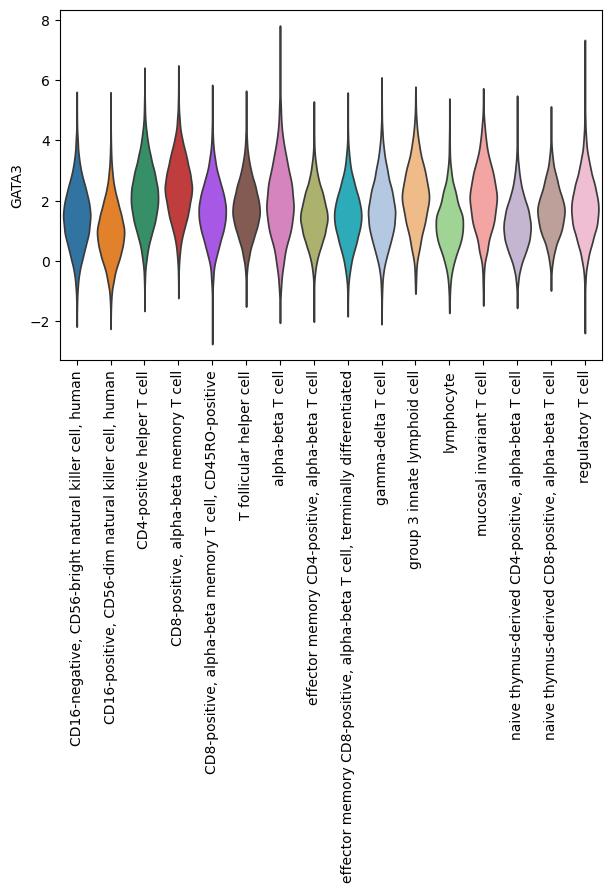
Transcription Factor Activity Inference with MLM#
Next, we infer transcription factor (TF) activity using the Multivariate Linear Model (MLM) from Decoupler.
Unlike ULM, which considers each TF independently, MLM accounts for multiple TFs simultaneously,
providing a more comprehensive estimation of regulatory influences.
%%time
rsc.dcg.mlm(data=adata, net=collectri, verbose = True, raw=False, bsize=10000, tmin=3)
CPU times: user 14.1 s, sys: 1.19 s, total: 15.3 s
Wall time: 15.2 s
Transcription Factor Activity Inference with AUCell#
Next, we infer transcription factor (TF) activity using AUCell from Decoupler.
AUCell estimates TF activity based on the enrichment of TF target genes within the top-expressed genes in each cell.
Unlike regression-based methods (ULM, MLM), AUCell is a rank-based approach that does not assume linear relationships.
%%time
rsc.dcg.aucell(data=adata, net=collectri, verbose = True, raw=False, bsize=5000, tmin=3)
CPU times: user 31.2 s, sys: 152 ms, total: 31.4 s
Wall time: 31.3 s
score = dc.pp.get_obsm(adata=adata, key="score_aucell")
sc.pl.embedding(score,basis="X_umap_harmony", color=["cell_type", "GATA3"], cmap='RdBu_r',ncols=1)
sc.pl.violin(score, keys=['GATA3'], groupby='cell_type', rotation=90, size = 0)


model = pd.read_parquet("nets/progeny.parquet")